
Genetic syndromes associated with Amenorrhea
Posted on June 27th 2016, by Dr. Anuradha Udumudi
Amenorrhoea is the absence of a menstrual period in a woman of reproductive age. Physiological states of amenorrhoea are seen most commonly, during pregnancy and lactation (breastfeeding). Amenorrhoea is a symptom with many potential causes. Primary amenorrhoea (menstruation cycles never starting) may be caused by developmental problems such as the congenital absence of the uterus, failure of the ovary to receive or maintain egg cells. It is defined as an absence of secondary sexual characteristics by age 14 with no menarche or normal secondary sexual characteristics but no menarche by 16 years of age. Secondary amenorrhoea (menstruation cycles ceasing) is often caused by hormonal disturbances from the hypothalamus and the pituitary gland, from premature menopause or intrauterine scar formation. It is defined as the absence of menses for three months in a woman with previously normal menstruation or nine months for women with a history of oligomenorrhoea. Oligomenorrhea is defined as menses occurring at intervals longer than 35 days apart.
Pathophysiology:
The menstrual cycle is an orderly progression of coordinated hormonal events in the female body that stimulates growth of a follicle to release an egg and prepare a site for implantation if fertilization should occur. The part of the brain called the hypothalamus regulates the menstrual cycle. The hypothalamus stimulates the pituitary gland. The pituitary gland lies just below the hypothalamus at the base of the brain. The pituitary gland releases two hormones that regulate the female reproductive cycle. They are luteinizing hormone (LH) and follicle-stimulating hormone (FSH). LH and FSH influence the production of estrogen and progesterone. These hormones control cyclic changes in the lining of the uterus. This includes menstruation. In order for a woman to have regular menstrual cycles, her hypothalamus, pituitary gland, ovaries and uterus must be functioning properly.
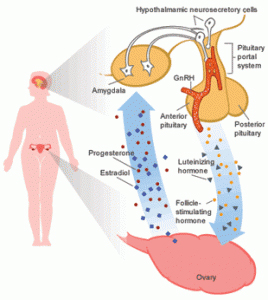
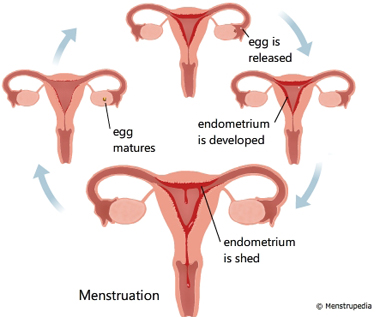
Figure: Hormonal regulation of Menstruation
Developmental and anatomical defects:
There is a range of developmental defects including agenesis, atresia, and septation of the reproductive tract, many of which have been associated with genetic syndromes. Several genes have been identified in the abnormal and normal development of the uterus, cervix, fallopian tubes and vagina. Many anomalies could be also multifactorial. But there are case reports of familial inheritance suggesting that specific genetic mutations may cause these defects. Furthermore, there are defined genetic syndromes that feature anomalies of the female reproductive tract.
Mullerian duct developmental and differentiation:
The urogenital system develops into the kidneys, gonads, and the urinary and reproductive tracts. The Wolffian (mesonephric) and Mullerian (paramesonephric) ducts are the primordia of the male and female reproductive tracts, respectively. Gonadal development is a separate developmental process that is determined by the sex chromosomes. Absence of anti-Müllerian hormone (AMH, normally produced by the male testes) will trigger stabilization of the Müllerian system and regression of the Wolffian system leading to development of the female reproductive tract. Development of the Müllerian ducts is considered a triphasic process consisting of initiation, invagination, and elongation.
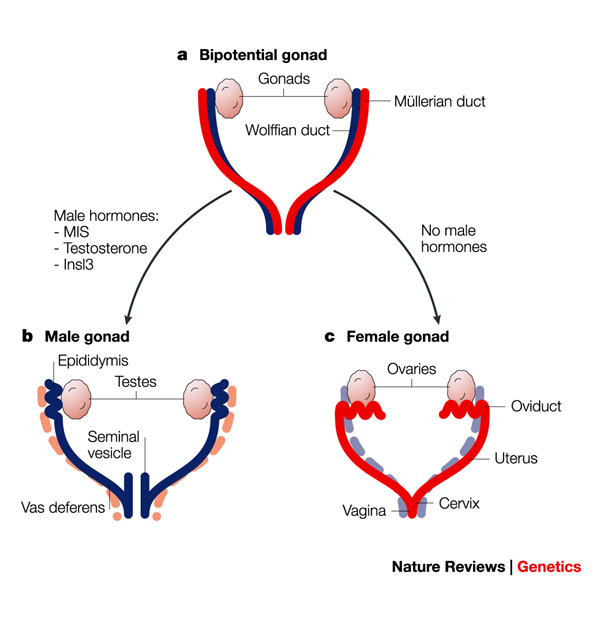
A set of genes play in an orchestral fashion to complete the Mullerian duct formation and differentiation. The genes involved are; Lim homeobox 1 (Lim1), Paired box 2 (Pax2), Wnt family (WNT4, Wnt9b, Wnt7a, Wnt5a), Empty spiracles homeobox 2 (Emx2), TCF2 and Dachshund homolog 1 and 2 (Dach1 and Dach2). Catenin (cadherin- associated protein), beta 1 (Ctnnb1) produces the protein β-catenin and is a downstream effector of the Wnt family genes.
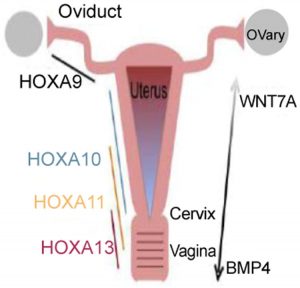
Following Müllerian duct formation, differentiation occurs along an antero-posterior (A-P) and radial axis. This includes the formation of the oviducts, uterus, cervix, and vagina. This occurs through interactions between the Müllerian duct epithelium and the surrounding mesenchyme. This A-P patterning establishes histologically distinct segmental boundaries. The anterior boundary occurs between the oviduct and uterine body, and the posterior boundary is between the uterus and cervix. This patterning is primarily regulated by Hoxa family homeobox transcription factors. Hoxa9, Hoxa10, Hoxa11, and Hoxa13 are expressed uniformly along the A-P axis. Hoxa9 is expressed in the oviduct whereas Hoxa10 and Hoxa11 are expressed in the uterus. Hoxa11 and Hoxa13 can be found in the cervix and anterior vagina. Hoxa10 and Hoxa11, as expected, are required for patterning and differentiation of the uterus and their expression patterns overlap during embryogenesis. Finally, forkhead box A2 (Foxa2) has been identified as an important regulatory gene in gland formation as ablation of Foxa2 leads to glandular agenesis.
Development of the Vagina:
It has been a commonly held belief (based on murine studies) that the vagina is of dual origin. The Müllerian ducts form the cranial portion, the so-called “Müllerian vagina”, while the urogenital sinus is the origin of the caudal portion, the so-called “sinus vagina” Vaginal atresia is a rare condition. In murine models, Hoxa13 and bone morphogenetic protein 4 (BMP4) appear to be strongly expressed in the Müllerian vagina but not in the uterus. The sinus vagina is also formed through the influence of BMP4 expression in the surrounding mesenchyme.
Genetic syndromes associated with Amenorhhea and female reproductive developmental defects
Amenorrhea could be due to chromosomal defects or single gene defects. Turner is the most common chromosomal condition.
1. Turner Syndrome:
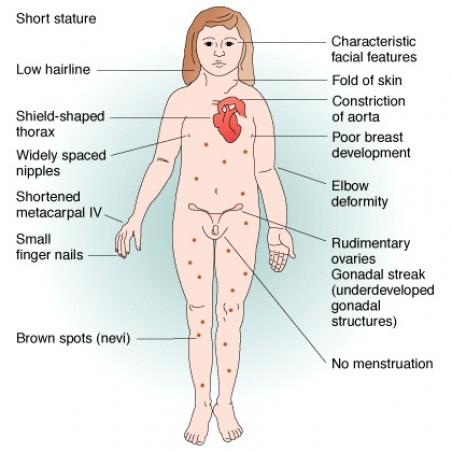
Turner syndrome is a chromosomal condition that affects development in females. This condition occurs in about 1 in 2,500 newborn girls worldwide, but it is much more common among pregnancies that do not survive to term (miscarriages and stillbirths). The most common feature of Turner syndrome is short stature, which becomes evident by about age 5. An early loss of ovarian function (ovarian hypofunction or premature ovarian failure) is also very common. The ovaries develop normally at first, but egg cells (oocytes) usually die prematurely and most ovarian tissue degenerates before birth. Many affected girls do not undergo puberty unless they receive hormone therapy, and most are unable to conceive (infertile). Most girls without Turner syndrome have their first menstrual period between 11 and 14 years old, so many girls with TS who have not started puberty choose to start estrogen treatments around that age, as their lack of breast development becomes more obvious. Even with years of estrogen replacement therapy it is rare for a woman with Turner syndrome to naturally conceive a child. This is because most women with TS have accelerated loss of eggs contained in the ovary.
A small percentage of females with Turner syndrome retain normal ovarian function through young adulthood. Some teenagers may undergo some breast development and begin menstruating, but cease further development and menses during the later teen years. A few women with Turner syndrome have apparently normal ovarian function with regular menses until the mid-20s before ovarian failure occurs.
Most women with Turner’s syndrome have ovarian dysgenesis; therefore, they are usually infertile, and in very rare cases have spontaneous menses followed by early menopause. Only 2% of the women have natural pregnancies, with high rates of miscarriages, stillbirths and malformed babies. Their pregnancy rate in oocyte donation programmes is 24-47%, but even these pregnancies have a high rate of miscarriage, probably due to uterine factors. The fertility options available for women with Turner syndrome are the same as those for women who also have reproductive issues but don’t have TS. These include unfertilized egg donations: Inserting an unfertilized egg from another woman into the fallopian tube or in vitro fertilization: placing a fertilized egg into the uterus
Phenotype:
Short stature: Growth rate in childhood is slightly slower; before age 11 years, some girls have height and growth rates that are well within the normal range, but heights are typically below the 50th percentile. The adolescent growth spurt is essentially absent
Puberty: Adrenarche, the beginning of pubic hair growth, occurs at a normal age. Breast development is absent when ovarian failure occurs before puberty. Primary or secondary amenorrhea occurs with ovarian failure.
Other characteristic physical findings:
-
Dental: A high arched palate, sometimes with dental crowding or malocclusion
-
Nails: Hypoplastic or hyperconvex nails
-
Nevi: Excessive numbers of nevi, when compared to other family members
-
Webbed neck: A broad neck and a low or indistinct hairline
-
Cubitus valgus (increased carrying angle)
-
Madelung deformities of the wrist
-
Short fourth and fifth metacarpals and metatarsals
-
Shield chest: The chest appears to be broad with widely spaced nipples
-
Lymphedema
-
Eyes: Ptosis, strabismus, amblyopia, and cataracts; epicanthal folds can be present; red-green color blindness
-
Ears: Serous otitis media is more common; the auricles may be posteriorly rotated or low set; hearing loss due to otosclerosis is common in adults
-
GI bleeding: This is usually due to intestinal vascular malformations, but the incidence of Crohn disease and ulcerative colitis is also increased
-
Scoliosis: This occurs in 10% of adolescents with Turner syndrome and may contribute to short stature
-
Hypertension: May be caused by coarctation of the aorta or renal anomalies but often occur even in the absence of such findings
-
Cardiac murmurs: Cardiovascular malformations include hypoplastic left heart, coarctation of the aorta, bicuspid aortic valve, and aortic dissection in adulthood
-
Thyroid: Hypothyroidism develops in 10-30% of patients and is often associated with thyroid enlargement
-
Cutis laxa: Loose folds of skin, particularly in the neck, are signs in newborns; this is a result of resolving lymphedema and occasionally is observed after infancy
Genetic cause:
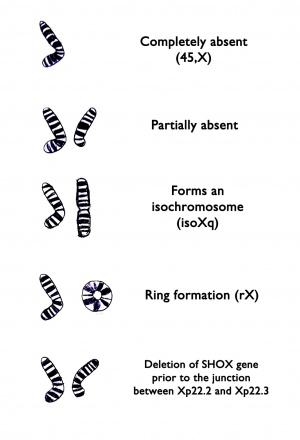
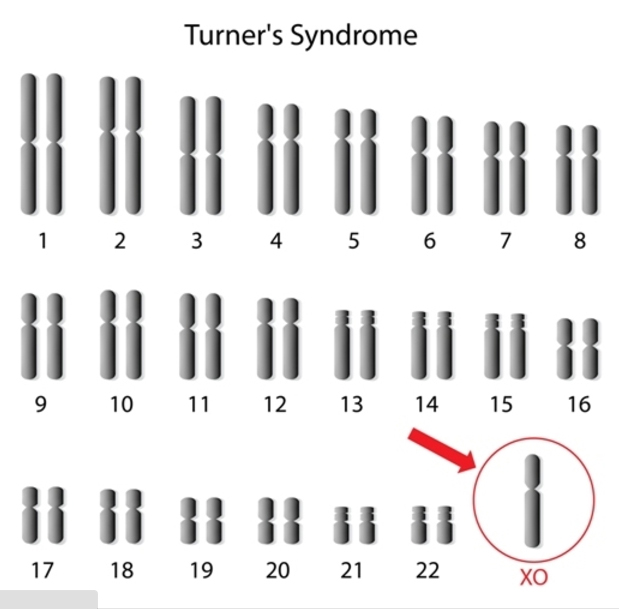
Turner syndrome results when one normal X chromosome is present in a female’s cells and the other sex chromosome is missing or structurally altered. The missing genetic material affects development before and after birth. About half of individuals with Turner syndrome have monosomy X. Turner syndrome can also occur if one of the sex chromosomes is partially missing or rearranged rather than completely absent. Some women with Turner syndrome have a chromosomal change in only some of their cells, which is known as mosaicism. Women with Turner syndrome caused by X chromosome mosaicism (45,X/46,XX or 45,X/46,XY) are said to have mosaic Turner syndrome.
Inheritance:
Most cases of Turner syndrome are not inherited. When this condition results from monosomy X, the chromosomal abnormality occurs as a random event during the formation of reproductive cells (eggs and sperm) in the affected person’s parent. An error in cell division called nondisjunction can result in reproductive cells with an abnormal number of chromosomes. For example, an egg or sperm cell may lose a sex chromosome as a result of nondisjunction. If one of these atypical reproductive cells contributes to the genetic makeup of a child, the child will have a single X chromosome in each cell and will be missing the other sex chromosome.
Mosaic Turner syndrome is also not inherited. In an affected individual, it occurs as a random event during cell division in early fetal development. As a result, some of an affected person’s cells have the usual two sex chromosomes, and other cells have only one copy of the X chromosome. Other sex chromosome abnormalities are also possible in females with X chromosome mosaicism. Rarely, Turner syndrome caused by a partial deletion of the X chromosome can be passed from one generation to the next.
Turner syndrome cytogenetic variants and their frequency is the following::
-
45x in 53% of cases of Turner syndrome
-
Mosaicism 45x / 46XX in 15% of cases of Turner syndrome
-
X isochromosome, 46X,i(Xq) in 10% of cases of Turner syndrome;
-
Mosacism 46X,i (Xq) / 46XX in 8% of cases of Turner syndrome;
-
Deletions 46XXp- or 46XXq- in 6% of cases of Turner syndrome;
-
Other mosaicism in 8% of cases of Turner syndrome.
Gene associated:
One copy of the SHOX gene is located on each of the sex chromosomes (the X and Y chromosomes). Because the SHOX gene is located on the sex chromosomes, most women with Turner syndrome have only one copy of the gene in each cell instead of the usual two copies. Loss of one copy of this gene reduces the amount of SHOX protein that is produced. A shortage of this protein likely contributes to the short stature and skeletal abnormalities (such as unusual rotation of the wrist and elbow joints) often seen in females with this condition. Cytogenetic Location: Xp22.33;Yp11.3, which is the short (p) arm of the X chromosome at position 22.33 and the short (p) arm of the Y chromosome at position 11.3
Inheritance
Turner syndrome is usually not inherited and is sporadic in most of the cases. Rarely, Turner syndrome caused by a partial deletion of the X chromosome can be passed from one generation to the next. Consanguinity, marraige between blood relatives has no role in having a Turner syndrome baby.
2. Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome:
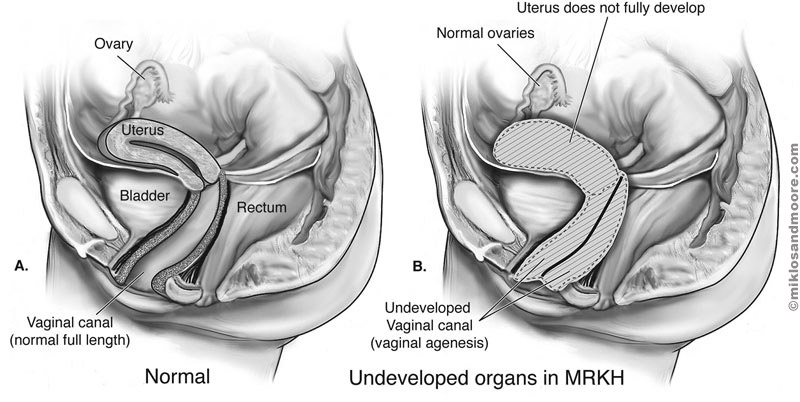
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is a disorder that occurs in females and mainly affects the reproductive system. Most often the diagnosis of Müllerian aplasia is labeled as MRKH syndrome. MRKH may feature isolated Müllerian aplasia (type 1), or be associated with renal, skeletal, auditory, and cardiac defects (type II).
Phenotype:
This condition causes the vagina and uterus to be underdeveloped or absent. Affected women usually do not have menstrual periods due to the absent uterus. Often, the first noticeable sign of MRKH syndrome is that menstruation does not begin by age 16 (primary amenorrhea). Women with MRKH syndrome have a female chromosome pattern (46,XX) and normally functioning ovaries. They also have normal female external genitalia and normal breast and pubic hair development. Although women with this condition are usually unable to carry a pregnancy, they may be able to have children through assisted reproduction. Women with MRKH syndrome may also have abnormalities in other parts of the body. The kidneys may be abnormally formed or positioned, or one kidney may fail to develop (unilateral renal agenesis). Affected individuals commonly develop skeletal abnormalities, particularly of the spinal bones (vertebrae). Females with MRKH syndrome may also have hearing loss or heart defects. MRKH syndrome affects approximately 1 in 4,500 newborn girls.
Gene:
The cause of MRKH syndrome is unknown, although it probably results from a combination of genetic and environmental factors. Researchers have not identified any genes associated with MRKH syndrome. The reproductive abnormalities of MRKH syndrome are due to incomplete development of the Müllerian duct.
Inheritance:
Most cases of MRKH syndrome occur in people with no history of the disorder in their family and is sporadic. Less often, MRKH syndrome is passed through generations in families. Its inheritance pattern is usually unclear because the signs and symptoms of the condition frequently vary among affected individuals in the same family. However, in some families, the condition appears to have an autosomal dominant pattern of inheritance.
3. Kallmann syndrome:
This is a condition characterized by delayed or absent puberty and an impaired sense of smell. At puberty, most affected individuals do not develop secondary sex characteristics, such as the growth of facial hair and deepening of the voice in males. This disorder is a form of hypogonadotropic hypogonadism (HH), which is a condition affecting the production of hormones that direct sexual development.
Phenotype:
At puberty, most affected individuals do not develop secondary sex characteristics, such as the growth of facial hair and deepening of the voice in males. Males with hypogonadotropic hypogonadism are often born with an unusually small penis (micropenis) and undescended testes (cryptorchidism). Affected females usually do not begin menstruating at puberty and have little or no breast development. In some people, puberty is incomplete or delayed.
In Kallmann syndrome, the sense of smell is either diminished (hyposmia) or completely absent (anosmia). This feature distinguishes Kallmann syndrome from most other forms of hypogonadotropic hypogonadism, which do not affect the sense of smell. Many people with Kallmann syndrome are not aware that they are unable to detect odors until the impairment is discovered through testing.
The features of Kallmann syndrome vary, even among affected people in the same family. Additional signs and symptoms can include a failure of one kidney to develop (unilateral renal agenesis), a cleft lip with or without an opening in the roof of the mouth (a cleft palate), abnormal eye movements, hearing loss, and abnormalities of tooth development. Some affected individuals have a condition called bimanual synkinesis, in which the movements of one hand are mirrored by the other hand. Bimanual synkinesis can make it difficult to do tasks that require the hands to move separately, such as playing a musical instrument.
Researchers have identified four forms of Kallmann syndrome, designated types 1 through 4, which are distinguished by their genetic cause. The four types are each characterized by hypogonadotropic hypogonadism and an impaired sense of smell. Additional features, such as a cleft palate, seem to occur only in types 1 and 2. Affected females usually do not begin menstruating at puberty and have little or no breast development. In some people, puberty is incomplete or delayed. Kallmann syndrome is estimated to affect 1 in 10,000 to 86,000 people and occurs more often in males than in females.
Gene:
Mutations in the ANOS1 ( Xp22.32), FGFR1 (8p11.23-p11.22), PROKR2 (20p12.3) , and PROK2 (3p13) genes cause Kallmann syndrome. Kallmann syndrome 1 (caused by ANOS1 gene mutations) has an X-linked recessive pattern of inheritance. Kallmann syndrome 2 results from mutations in the FGFR1 gene. Mutations in the PROKR2 and PROK2 genes cause Kallmann syndrome types 3 and 4, respectively.
The ANOS1, FGFR1, PROKR2, and PROK2 genes play a role in the migration of neurons that produce a hormone called gonadotropin-releasing hormone (GnRH). These hormones are important for the normal function of the gonads (ovaries in women and testes in men). Together, mutations in the ANOS1, FGFR1, PROKR2, and PROK2 genes account for 25 percent to 30 percent of all cases of Kallmann syndrome. In cases without an identified mutation in one of these genes, the cause of the condition is unknown.
Inheritance:
Kallmann syndrome 1 (caused by ANOS1 gene mutations) has an X-linked recessive pattern of inheritance. Most cases of Kallmann syndrome 1 are described as simplex, which means only one person in a family is affected. Some affected people inherit a ANOS1 gene mutation from their mothers, who carry a single mutated copy of the gene in each cell. Other people have the condition as a result of a new mutation in the ANOS1 gene.
Other forms of Kallmann syndrome can be inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.
In several families, Kallmann syndrome has shown an autosomal recessive pattern of inheritance. Mutations in at least 20 genes cause hypogonadotropic hypogonadism including Kallmann syndrome in about 35–40% of patients.
4. Perrault syndrome:
It is characterized by sensorineural hearing loss (SNHL) in males and females, and ovarian dysfunction in females. When onset is in early childhood, hearing loss can be progressive. Ovarian dysfunction ranges from gonadal dysgenesis (absent or streak gonads) manifesting as primary amenorrhea to primary ovarian insufficiency (POI) defined as cessation of menses before age 40 years.
Phenotype:
In Perrault syndrome, the problems with hearing are caused by changes in the inner ear, which is known as sensorineural hearing loss. The impairment usually affects both ears and can be present at birth or begin in early childhood. Unless hearing is completely impaired at birth, the hearing problems worsen over time.
Females with Perrault syndrome have abnormal or missing ovaries (ovarian dysgenesis), although their external genitalia are normal. Severely affected girls do not begin menstruation by age 16 (primary amenorrhea), and most never have a menstrual period. Less severely affected women have an early loss of ovarian function (primary ovarian insufficiency); their menstrual periods begin in adolescence, but they become less frequent and eventually stop before age 40. Women with Perrault syndrome may have difficulty conceiving or be unable to have biological children (infertile).
Neurological problems in individuals with Perrault syndrome can include intellectual disability, difficulty with balance and coordinating movements (ataxia), and loss of sensation and weakness in the limbs (peripheral neuropathy). However, not everyone with this condition has neurological problems.
The diagnosis of Perrault syndrome is based on the clinical findings of SNHL in men and women, and ovarian dysfunction in women with a 46,XX karyotype. Perrault syndrome is a rare disorder; fewer than 100 affected individuals have been described in the medical literature.
Gene:
Perrault syndrome has several genetic causes. C10orf2 (10q24), CLPP ( 19p13.3), HARS2 (5q31.3), LARS2 (3p21.3), or HSD17B4 (5q21) gene mutations have been found in a small number of affected individuals. The proteins produced from several of these genes, including C10orf2, CLPP, HARS2, and LARS2, function in cell structures called mitochondria, which convert the energy from food into a form that cells can use. Although the effect of these gene mutations on mitochondrial function is unknown, researchers speculate that disruption of mitochondrial energy production could underlie the signs and symptoms of Perrault syndrome.
The protein produced from the HSD17B4 gene is active in cell structures called peroxisomes, which contain a variety of enzymes that break down many different substances in cells. It is not known how mutations in this gene affect peroxisome function or lead to hearing loss in affected males and ovarian abnormalities in females with Perrault syndrome.
It is likely that other genes that have not been identified are also involved in this condition.
Inheritance:
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they do not show signs and symptoms of the condition.
5. McKusick-Kaufman (MKKS):
McKusick-Kaufman syndrome is a condition that affects the development of the hands and feet, heart, and reproductive system.
Phenotype:
It is characterized by a combination of three features: extra fingers and/or toes (polydactyly), heart defects, and genital abnormalities. This condition was first described in the Old Order Amish population, where it affects an estimated 1 in 10,000 people. The incidence of McKusick-Kaufman syndrome in non-Amish populations is unknown.
Most females with McKusick-Kaufman syndrome are born with a genital abnormality called hydrometrocolpos, which is a large accumulation of fluid in the pelvis. Neonatal hydrometrocolpos is rare Mullerian duct anomaly caused by obstruction of the vagina. Hydrometrocolpos results from a blockage of the vagina before birth, which can occur if part of the vagina fails to develop (vaginal agenesis) or if a membrane blocks the opening of the vagina. This blockage allows fluid to build up in the vagina and uterus, stretching these organs and leading to a fluid-filled mass. Though MKKS is rare in males, if present, it is sometimes associated with hypospadias, chordee, and cryptorchidism.
MKKS shows association with congenital heart defects such as atrioventricular canal defects, ventricular septal defect, and hypoplastic left heart from 10% to 20% of cases. Other less commonly associated findings are gastrointestinal abnormalities (28%) that consist of imperforate anus, rectovaginal or vesicovaginal fistula, Hirschsprung’s disease, and malrotation. Abnormalities of the eyes (5%).
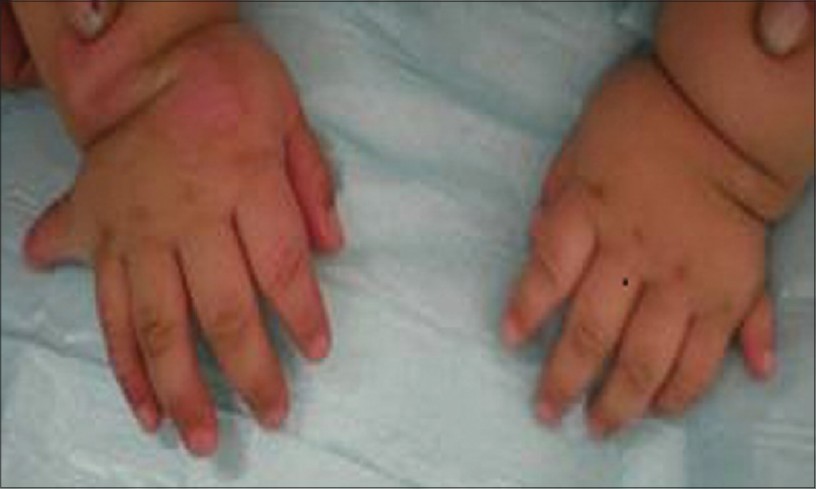
Fig: Postaxial polydactyly
Gene:
Mutations in the MKKS gene cause McKusick-Kaufman syndrome. This gene provides instructions for making a protein that plays an important role in the formation of the limbs, heart, and reproductive system. The protein’s structure suggests that it may act as a chaperonin, which is a type of protein that helps fold other proteins. Although the structure of the MKKS protein is similar to that of a chaperonin, some recent studies have suggested that protein folding may not be this protein’s primary function. The mutations that underlie McKusick-Kaufman syndrome alter the structure of the MKKS protein. Although the altered protein disrupts the development of several parts of the body before birth, it is unclear how MKKS mutations lead to the specific features of this disorder.
Cytogenetic Location: 20p12, which is the short (p) arm of chromosome 20 at position 12.
Inheritance:
This condition is inherited in an autosomal recessive pattern.
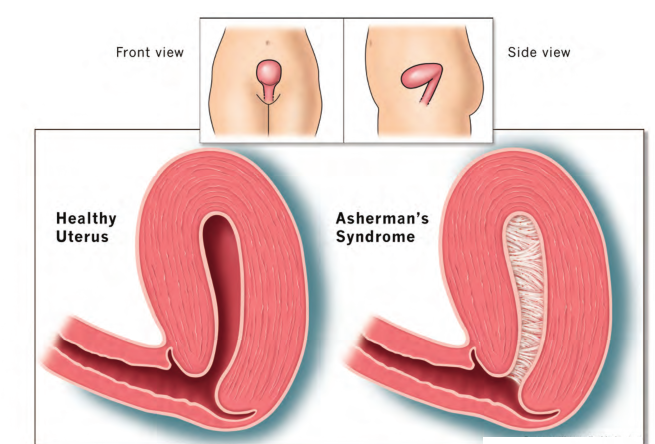
6. Asherman’s syndrome:
Asherman’s syndrome (AS) or Fritsch syndrome, is a condition characterized by adhesions and/or fibrosis of the endometrium particularly but can also affect the myometrium. It is often associated with dilation and curettage of the intrauterine cavity.
A number of other terms have been used to describe the condition and related conditions including: intrauterine adhesions (IUA), uterine/cervical atresia, traumatic uterine atrophy, sclerotic endometrium, endometrial sclerosis, and intrauterine synechiae. Often, patients experience secondary menstrual irregularities characterized by a decrease in flow and duration of bleeding (amenorrhea, hypomenorrhea, or oligomenorrhea) and become infertile. Menstrual anomalies are often but not always correlated with severity: adhesions restricted to only the cervix or lower uterus may block menstruation. Pain during menstruation and ovulation is sometimes experienced and can be attributed to blockages.
7. Other associated syndromes:
-
Johanson-Blizzard syndrome has been associated with multiple anomalies including longitudinal vaginal septa. It has been identified as ubiquitin protein ligase E3 component n-recognin 1 (UBR1), but the genetic mechanisms leading to the known phenotype are not fully understood as the gene is generally known to be involved in a proteolytic pathway of the ubiquitin system.
-
DeCherney syndrome is a case of bladder exstrophy with an upward anteriorly displaced bicornuate uterus and an associated incomplete transverse vaginal septum. The gene for a third syndrome, hand-foot-genital syndrome is characterized by hand defects, urinary tract anomalies, and Müllerian ducts anomalies. Absence of the labia, has been reported in popliteal pterygium syndrome. In humans the genetic syndrome of ectrodactyly, ectodermal dysplasia, and facial clefts (ECC) results from p63 mutations.
-
Ulnar-mammary syndrome is associated with upper limb structures, apocrine/mammary hypoplasias, dental abnormalities, and genital anomalies including imperforate hymen. Alterations in the TBX3 gene, a downstream target of retinoic acid, have been implicated as the cause of ulnar-mammary syndrome.
-
WNT7A mutations have recently been recognized as causing Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome; which is characterized as having several limb deformities and uterine hypoplasias/aplasia.
-
Duplication in the SHOX gene has recently been identified in two daughters with MRKH type I and their phenotypically normal father.
-
Microdeletions in 1q21.1 have also been found most often associated with thrombocytopenia-absent radius syndrome (TAR).
-
The incidence of uterine and genital abnormalities is estimated to be 6% of cases of TAR syndrome.
The above chromosome loci and genes discussed may partially explain the familial and syndromic cases of Müllerian aplasia; however, most cases remain sporadic. Understanding the underlying genetic mechanisms of abnormal development of the female reproductive tract may help us understand these adverse outcomes.
References:
1. Genetic Syndromes and Genes Involved in the Development of the Female Reproductive Tract: A Possible Role for Gene Therapy. MT Connell, CM Owen and JH Segars, J Genet Syndr Gene Ther (2013); 4, 1-24.
2. Turner Syndrome and its Variants. R. Bharath, A.G Unnikrishnan, M.V Thampy1, Alka Anilkumar1, B Nisha, V.P Praveen, Vasantha Nair, R.V. Jayakumar and Harish Kumar, Indian Journal of Pediatrics (2010); 77, 193-195.
3. The genetic basis of female reproductive disorders: Etiology and clinical testing. Lawrence C. Layman, Mol Cell Endocrinol. (2013) 370(0), 138–148.
Note: Content copyrighted by GeneTech. Please share link to this page freely. Quote with proper attribution. Do not copy partial or full content without attribution. Contact us if you have any questions.