
Molecular Genetics of Beta Thalassemia
Posted on May 16th 2016, by Dr. Anuradha Udumudi
GENETICS FOR PHYSICIANS SERIES
Beta-thalassemia is one of the most common autosomal recessive disorders worldwide with high prevalence is Indian sub continent. It is caused by the reduced (beta+) or absent (beta0) synthesis of the beta globin chains of the hemoglobin (Hb) tetramer, which is made up of two alpha globin and two beta globin chains (alpha2beta2). When the beta globin chains are reduced or absent, the unassembled alpha chains precipitate and lead to oxidative damage of the cell membrane, thereby resulting in apoptosis (ineffective erythropoiesis).
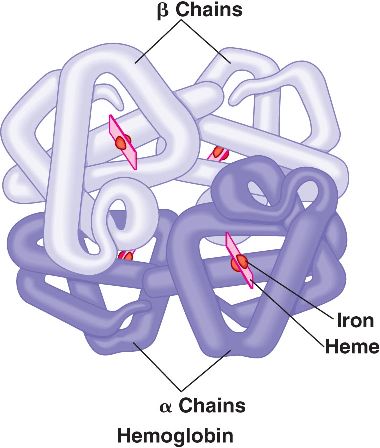
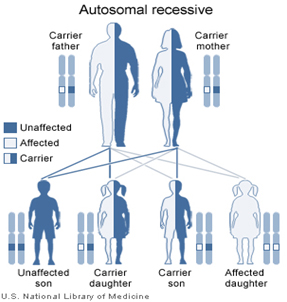
Mutations on beta globin gene (HBB) cause thalassemia when present in homozygous condition (mutations present on both copies of genes). It inherits in autosomal recessive pattern. Normal parents who carry mutation in a heterozygous condition (mutation on only one copy of gene) have 25% risk of giving birth to a child with Thalassemia.
Homozygotes for beta-thalassemia mutation may develop either thalassemia major or thalassemia intermedia. Thalassemia Major is a severe transfusion-dependent anemia. Thalassemia intermedia comprehends a clinically and genotypically very heterogeneous group of thalassemia-like disorders, ranging in severity from the asymptomatic carrier state to the severe transfusion-dependent type. The clinical severity of beta-thalassemia is related to the extent of imbalance between the alpha globin and non-alpha globin chains.
Clinical Beta Thalassemia major and Intermedia
Affected infants with thalassemia major fail to thrive and become progressively pale. Feeding problems, diarrhea, irritability, recurrent bouts of fever, and enlargement of the abdomen, caused by splenomegaly, may occur. If a regular transfusion program that maintains a minimum Hb concentration of 95–105 g/L is initiated, then growth and development are normal until the age of 10–11 years. After the age of 10–11 years, affected individuals are at risk of developing severe complications related to posttransfusional iron overload, depending on their compliance with chelation therapy.
Complications can include growth retardation and failure of sexual maturation, HFE-associated hereditary hemochromatosis, dilated myocardiopathy and pericarditis, chronic hepatitis, fibrosis, and cirrhosis, diabetes mellitus and Infectious complications, including hepatitis B and C virus and HIV. Other complications are hypersplenism (usually related to late and irregular transfusions), venous thrombosis (occurring especially after splenectomy), osteoporosis (which is multifactorial).
The most relevant features of untreated or poorly transfused individuals are growth retardation, pallor, jaundice, brown pigmentation of the skin, poor musculature, genu valgum, hepatosplenomegaly, leg ulcers, development of masses from extramedullary hematopoiesis, and skeletal changes that result from expansion of the bone marrow.
Patients with thalassemia intermedia show a markedly heterogeneous clinical picture. Principle symptoms are pallor, jaundice, cholelithiasis, liver and spleen enlargement, moderate to severe skeletal changes, leg ulcers, extramedullary masses of hyperplastic erythroid marrow, a tendency to develop osteopenia and osteoporosis, and thrombotic complications resulting from a hypercoagulable state. By definition, transfusions are not required or only occasionally required. Iron overload occurs mainly from increased intestinal absorption of iron caused by ineffective erythropoiesis.
Patients with thalassemia major have a severe microcytic and hypochromic anemia, associated with increased number of red blood cells and low mean corpuscular volume (MCV) and mean corpuscular Hb (MCH). Peripheral blood smear shows, in addition to microcytosis and hypochromia, anisocytosis, poikilocytosis (spiculated tear drop and elongated cells), and nucleated red blood cells (i.e., erythroblasts). The number of erythroblasts is related to the degree of anemia and is markedly increased after splenectomy. Hb pattern (by HPLC) varies with HbA absent, HbF 95–98%, and HbA2 2–5%. Patients with thalassemia intermedia have a moderate anemia and show a markedly heterogeneous hematological picture, ranging in severity from that of the beta-thalassemia carrier state to that of thalassemia major.
Sickle cell disease is a variant form having atypical hemoglobin molecules called hemoglobin S, which can distort red blood cells into a sickle, or crescent, shape.
A comprehensive management of thalassemia major and thalassemia intermedia includes,transfusion program, management of iron overload, use of blood chelators and bone marrow transportation.
Beta-thalassemia carrier state
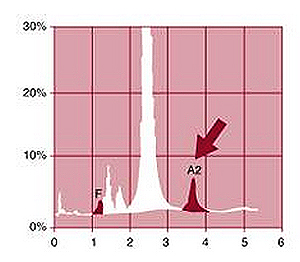
Hetrozygous mutations on beta globin gene are seen in carriers of beta thalassemia. Carriers of beta-thalassemia are clinically asymptomatic. The characteristic hematological features are microcytosis (reduced red blood cell volume) and hypochromia (reduced red blood cell Hb content). The Hb pattern (by HPLC) of beta-thalassemia heterozygotes is characterized by 92–95% HbA, >3.8 HbA2, and variable amount of HbF (0.5–4%). Examination of the blood smear reveals microcytosis, hypochromia, and marked variations in size and shape of the red blood cells.
Gene mutations and Phenotype/Genotype correlation
Beta Thalassemia is caused by beta globin (HBB) gene that maps on to the short arm of chromosome 11 (location 11p15.5). More than 200 disease-causing mutations have been so far identified. Mutations like deletion, initiation codon, nonsense, frameshift, and splicing mutations, especially at the splice-site junction cause severe phenotype. On the other hand, mutations in the promoter area, polyadenylation signal, 5′ or 3′ UTR and splicing abnormalities result in reduced production of beta chains. Despite the marked molecular heterogeneity, the prevalent molecular defects are limited in each at risk population, in which 4–10 mutations usually account for most of the beta globin disease-causing allele.
Homozygosity or compound heterozygosity for beta-thalassemia most commonly result in the clinical phenotype of transfusion-dependent thalassemia major. However, a consistent proportion of homozygotes develop milder forms, called thalassemia intermedia, which range in severity from thalassemia major to the beta-thalassemia carrier state
Phenotype and genotype correlation involves a complex mechanism of coinheritance with alpha thalassemia mutations and genetic determinants causing persistant increase of HbF. Recent studies indicate that the BCL11A, HBS1L-MYB region, and alpha-thalassemia contribute differently in the amelioration of clinical severity in thalassemia intermedia. However, despite the significant progress made in this field of thalassemia disorders, in a significant number of cases of thalassemia intermedia, the molecular mechanism has not been so far elucidated.
In rare instances, the beta-thalassemia defect does not lie in the beta globin gene cluster. Some mutations in the X-linked transcription factor GATA-1 may produce thrombocytopenia associated with thalassemia trait, and defects in transcription factor TFIIH results in thalassemia like hematolocical phenotype in addition to xeroderma pigmentosum and trichothiodystrophy.
The Indian scenario
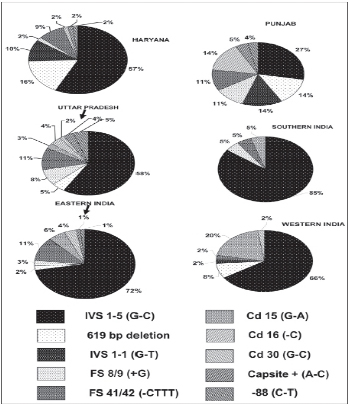
β-thalassemia is the commonest single-gene disorder in the Indian population. Ten percent of the total world thalassemics are born in India every year. It has been estimated that the prevalence of pathological hemoglobinopathies in India is 1.2/1,000 live births with approximately 32,400 babies with a serious hemoglobin disorder. The five common mutations on beta globin gene -IVS 1-5 G → C, IVS 1 -1 G → T, Codon 41/42 (- TCTT), Codon 8/9 and the 619 bp deletion account for over 90% of the mutations in beta thalassemia patients.
Beta Thalassemia Carrier detection
Because of the high carrier rate for HBB mutations in Indian population, it is important to screen population for carrier status. One of the best way to prevent thalassemia is to screen pregnant woman for thalassemia carrier status. This can be achieved by Hb chromatography by HPLC, which can quantitate HbA2 and HbF and can detect the most common Hb variants (HbS, HbC, and HbE) that may result in a Hb disorder by interacting with beta-thalassemia.
If the pregnant woman is screened positive, partner can be screened. If both the partners are known to be carriers based on the screening, genetic counseling along with molecular diagnosis must be offerd to couple. If both partners of a couple have the HBB disease-causing mutation, each of their offspring has a ¼ risk of being affected. Prenatal diagnosis is recommended if the thalassemia mutation is identified in the carrier parent or in an affected family member (proband).
The optimal time for the assessment of genetic risk, definition of carrier status, and genetic counseling is before pregnancy. Since beta thalassemia is an autosomal recessive disorder, chances for partner carrier status is more in consanguineous marriages. It is appropriate to offer genetic counseling (including discussion on the availability of prenatal diagnosis, potential risks to offspring, and reproductive options) to young adults who are carriers.
Population screening associated with genetic counseling is extremely useful by allowing couples at risk to make informed decision on their reproductive choices. Furthermore, in the population at risk targeted by screening will result in consistent reduction of the birth rate of affected children.
Molecular diagnosis of beta-thalassemia
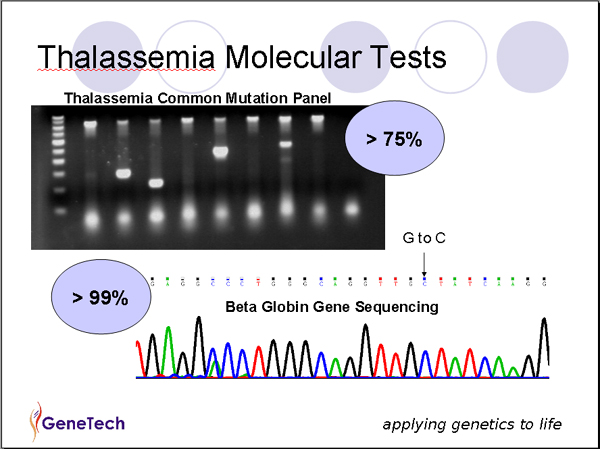
Commonly occurring mutations of the HBB gene are detected by a number of polymerase chain reaction (PCR)-based procedures. If targeted mutation analysis fails to detect common mutations, full gene sequencing of beta globin gene can be performed. Sensitivity of the tests is 99%. Presence of an extended deletion should be investigated by using multiplex ligation-dependent probe amplification (MPLA).
Prenatal diagnosis
In high-risk pregnancies in which both members are defined carriers for beta-thalassemia, prenatal diagnosis is possible by analysis of DNA extracted from fetal cells. Chorionic villus sampling is a procedure performed at 10-13 weeks of gestation whereas amniocentesis is usually performed at approximately 15–18 weeks’ gestation.
References
-
Beta-thalassemia. Antonio Cao and Renzo Galanello Genetics in Medicine (2010) 12, 61–76; doi:10.1097/GIM.0b013e3181cd68ed
- Beta Thalassemia in India: Current status and the challenges ahead. Kirti Grow, Minakshi Vashist, Pankaj Abrol, Siksha Sharma, Ritu Yadav. International Journal of Pharmacy and Pharmaceutical Sciences. ISSN- 0975-1491 Vol 6, Issue 4, 2014
- Mutational spectrum of thalassemias in India. Inusha Panigrahi, RK Marwaha. Division of Genetics, Department of Pediatrics, Advanced Pediatric Center, Postgraduate Institute of Medical Education and Research (PGIMER), Chandigarh, India
Note: Content copyrighted by GeneTech. Please share link to this page freely. Quote with proper attribution. Do not copy partial or full content without attribution. Contact us if you have any questions.